NPs Basic Information
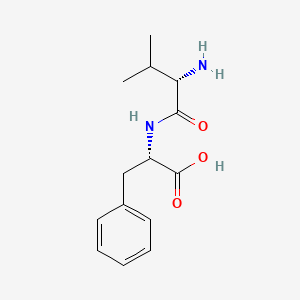
|
Name |
L-valyl-L-phenylalanine
|
| Molecular Formula | C14H20N2O3 | |
| IUPAC Name* |
(2S)-2-[[(2S)-2-amino-3-methylbutanoyl]amino]-3-phenylpropanoic acid
|
|
| SMILES |
CC(C)[C@@H](C(=O)N[C@@H](CC1=CC=CC=C1)C(=O)O)N
|
|
| InChI |
InChI=1S/C14H20N2O3/c1-9(2)12(15)13(17)16-11(14(18)19)8-10-6-4-3-5-7-10/h3-7,9,11-12H,8,15H2,1-2H3,(H,16,17)(H,18,19)/t11-,12-/m0/s1
|
|
| InChIKey |
GJNDXQBALKCYSZ-RYUDHWBXSA-N
|
|
| Synonyms |
H-VAL-PHE-OH; 3918-92-1; L-valyl-L-phenylalanine; valylphenylalanine; VAL-PHE; L-Phenylalanine, L-valyl-; CHEMBL8486; CHEBI:75016; (2S)-2-[(2S)-2-amino-3-methylbutanamido]-3-phenylpropanoic acid; (2S)-2-[[(2S)-2-amino-3-methylbutanoyl]amino]-3-phenylpropanoic acid; (2S)-2-[[(2S)-2-azaniumyl-3-methylbutanoyl]amino]-3-phenylpropanoate; L-Val-L-Phe; VF dipeptide; V-F Dipeptide; Valyl-Phenylalanine; L-Val-L-Phe-OH; Valine Phenylalanine dipeptide; Valine-Phenylalanine dipeptide; SCHEMBL7335375; DTXSID80959966; 75946-40-6; ZINC1605725; BDBM50142287; MFCD00020423; AKOS010421061; CS-7910; BS-49068; DS-012099; HY-107378; E78082; EN300-16256474; Q27145075; (S)-2-((S)-2-Amino-3-methyl-butyrylamino)-3-phenyl-propionic acid
|
|
| CAS | 3918-92-1 | |
| PubChem CID | 6993120 | |
| ChEMBL ID | CHEMBL8486 |
*Note: the IUPAC Name was collected from PubChem.
Chemical Classification: |
|
|
|---|
——————————————————————————————————————————
NPs Species Source
| Endophyte ID | Endophyte Name | Family | Genus | Taxonomy ID | GenBank ID | Closest GenBank ID | Reference | |
|---|---|---|---|---|---|---|---|---|
| Endophyte ID | Endophyte Name | Family | Genus | Taxonomy ID | GenBank ID | Closest GenBank ID | Reference |
NPs Biological Activity
| Bioactivity Name | Target ID | Target Name | Target Type | Target Organism | Target Organism ID | Potency of Bioactivity | Activity Type | Value | Unit | Endophyte ID | Endophyte Name | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Bioactivity Name | Target ID | Target Name | Target Type | Target Organism | Target Organism ID | Potency of Bioactivity | Activity Type | Value | Unit | Endophyte ID | Endophyte Name |
NPs Physi-Chem Properties
| Molecular Weight: | 264.32 | ALogp: | -2.2 |
| HBD: | 3 | HBA: | 4 |
| Rotatable Bonds: | 6 | Lipinski's rule of five: | Accepted |
| Polar Surface Area: | 92.4 | Aromatic Rings: | 1 |
| Heavy Atoms: | 19 | QED Weighted: | 0.719 |
——————————————————————————————————————————
NPs ADMET Properties*
ADMET: Absorption
| Caco-2 Permeability: | -6.05 | MDCK Permeability: | 0.00105773 |
| Pgp-inhibitor: | 0 | Pgp-substrate: | 0.012 |
| Human Intestinal Absorption (HIA): | 0.013 | 20% Bioavailability (F20%): | 0.002 |
| 30% Bioavailability (F30%): | 0.001 |
——————————————————————————————————————————
ADMET: Distribution
| Blood-Brain-Barrier Penetration (BBB): | 0.829 | Plasma Protein Binding (PPB): | 31.53% |
| Volume Distribution (VD): | 0.254 | Fu: | 64.84% |
——————————————————————————————————————————
ADMET: Metabolism
| CYP1A2-inhibitor: | 0.034 | CYP1A2-substrate: | 0.04 |
| CYP2C19-inhibitor: | 0.069 | CYP2C19-substrate: | 0.084 |
| CYP2C9-inhibitor: | 0.025 | CYP2C9-substrate: | 0.56 |
| CYP2D6-inhibitor: | 0.146 | CYP2D6-substrate: | 0.213 |
| CYP3A4-inhibitor: | 0.029 | CYP3A4-substrate: | 0.164 |
——————————————————————————————————————————
ADMET: Excretion
| Clearance (CL): | 4.218 | Half-life (T1/2): | 0.872 |
——————————————————————————————————————————
ADMET: Toxicity
| hERG Blockers: | 0.03 | Human Hepatotoxicity (H-HT): | 0.33 |
| Drug-inuced Liver Injury (DILI): | 0.101 | AMES Toxicity: | 0.009 |
| Rat Oral Acute Toxicity: | 0.5 | Maximum Recommended Daily Dose: | 0.02 |
| Skin Sensitization: | 0.078 | Carcinogencity: | 0.052 |
| Eye Corrosion: | 0.004 | Eye Irritation: | 0.03 |
| Respiratory Toxicity: | 0.166 |
——————————————————————————————————————————
*Note: the ADMET properties was calculated by ADMETlab 2.0. Reference: PMID: 33893803.
Similar Compounds*
Compounds similar to EMNPD with top10 similarity:
| Similar NPs | Similar Drugs | ||||||
|---|---|---|---|---|---|---|---|
| NPs ID | NPs 2D Structure | Similarity Score | TTD ID | Drug 2D Structure | Similarity Score | ||
| ENC000717 | 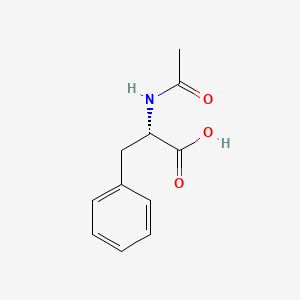 |
0.673 | D0RA5Q |  |
0.667 | ||
| ENC002126 | 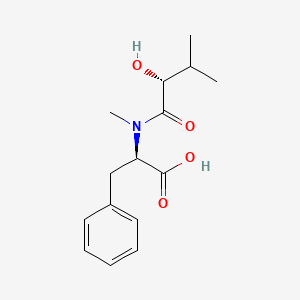 |
0.552 | D06PSS | 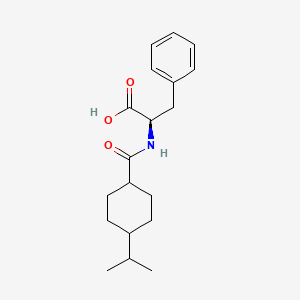 |
0.554 | ||
| ENC000130 | 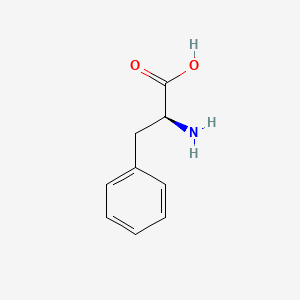 |
0.527 | D0R1CR | 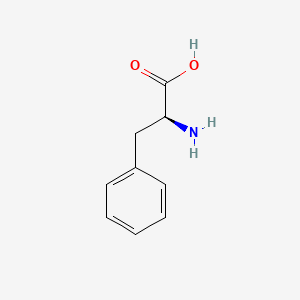 |
0.527 | ||
| ENC001819 | 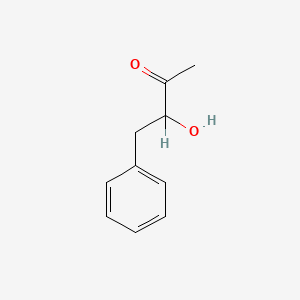 |
0.448 | D05BMG | 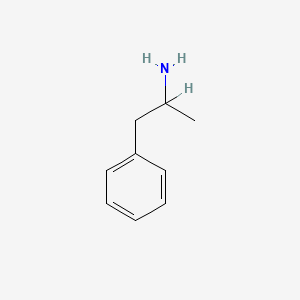 |
0.463 | ||
| ENC001514 | 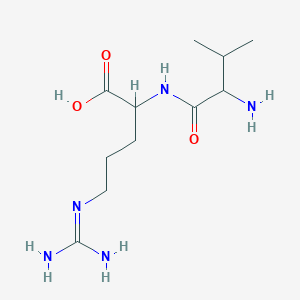 |
0.429 | D0T3LF | 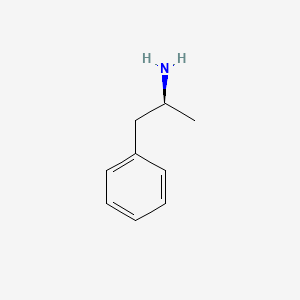 |
0.463 | ||
| ENC000214 | 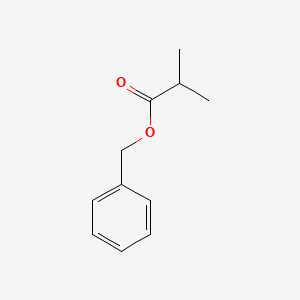 |
0.426 | D0X5SJ | 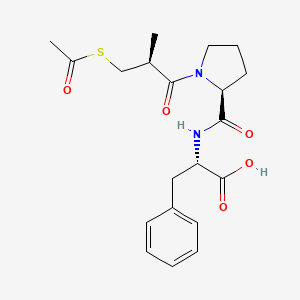 |
0.455 | ||
| ENC001906 | 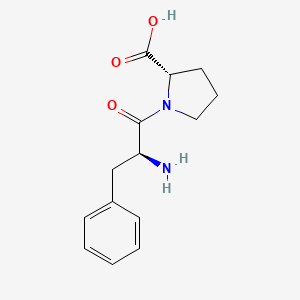 |
0.425 | D00DEF |  |
0.444 | ||
| ENC001902 | 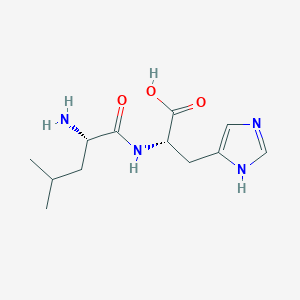 |
0.417 | D05OFX | 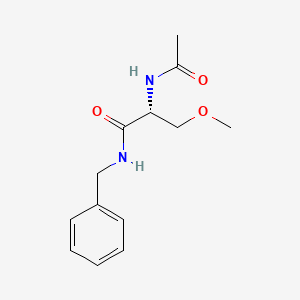 |
0.443 | ||
| ENC000054 | 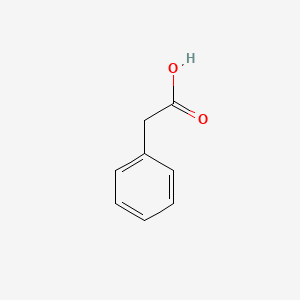 |
0.411 | D0P6UB |  |
0.439 | ||
| ENC002451 |  |
0.410 | D0SH3I |  |
0.433 | ||